


















Learn how we've leveraged funding and partnerships to help Ontario's regenerative medicine ecosystem thrive.
OIRM propels Ontario-made research toward the clinic, playing a key role in 22 clinical trials to date.

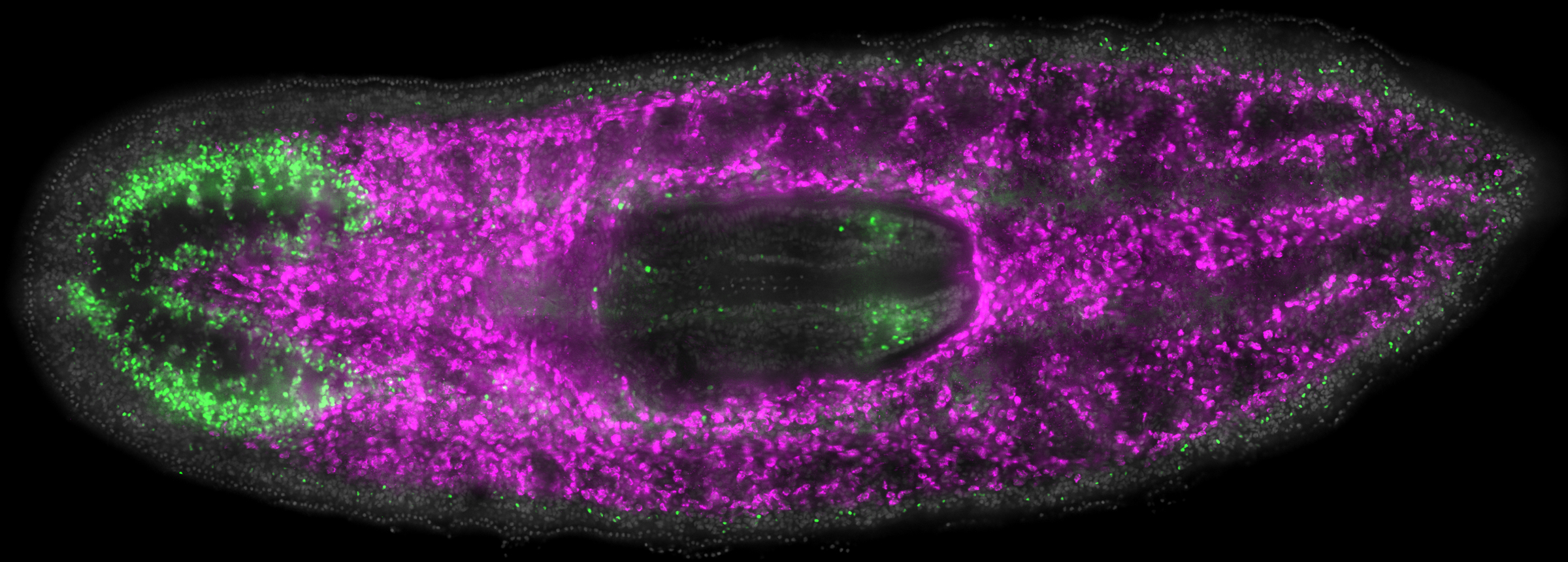
Go in-depth on the research our teams are working on, and the complex scientific and clinical challenges they are trying to solve.
Our world-class innovators collaborate locally and internationally to advance the development and commercialization of stem cell therapies.
OIRM, C3i Center Inc. and other partners are ensuring made-in-Ontario technologies get the strategic support they need to succeed.
We're committed to help research progress from bench to bedside by putting the right partnerships in place to create a streamlined infrastructure.